We just solved a problem that I never wanted to solve: That is, transferable retention time prediction of small molecules. They say that the best king is someone who does not want to be king; so maybe, the best problem solver is someone who does not want to solve the problem? Not sure about that. (My analogies have been quite monarchist lately; we are watching too much The Crown.)
What is transferable retention time prediction? Let’s assume you tell me some details about your chromatographic setup. We are talking about liquid, reversed-phase chromatography. You tell me what column you use, what gradient, what pH, maybe what temperature. Then, you say “3-Ketocholanic acid” and I say, “6.89 min”. You say “Dibutyl phthalate”, I say “7.39 min”. That’s it, folks. If you think that is not possible: Those are real-world examples from a biological dataset. The true (experimentally measured) retention times where 6.65 min and 7.41 min.
Now, you might say, “but how many authentic standards were measured on the same chromatographic setup so that you could do your predictions”? Because that is how retention time prediction works, right? You need authentic standards measured on the same system so that you can do predictions, right? Not for transferable retention time prediction. For our method, the answer to the above question is zero, at least in general. For the above real-world example, the answer is 19. Nineteen. In detail, 19 NAPS (N-Alkylpyridinium 3-sulfonate) molecules were measured in an independent run. Those standards were helpful, but we could have done without them. If you know a bit about retention time prediction: There was no fine-tuning of some machine learning model on the target dataset. No, sir.
Now comes the cool part: Our method for retention time prediction already performs better than any other method for retention time prediction. The performance is almost as good as that of best-in-class methods if we do a random (uniform) split of the data on the target system. Yet, we all know (or, we all should know) that uniform splitting of small molecules is not a smart idea; it results in massively overestimating the power of machine learning models. Now, our model is not trained (fine-tuned) on data from the target system. Effect: Our method works basically equally well for any splitting of the target dataset. And, we already outperform the best-in-class method.
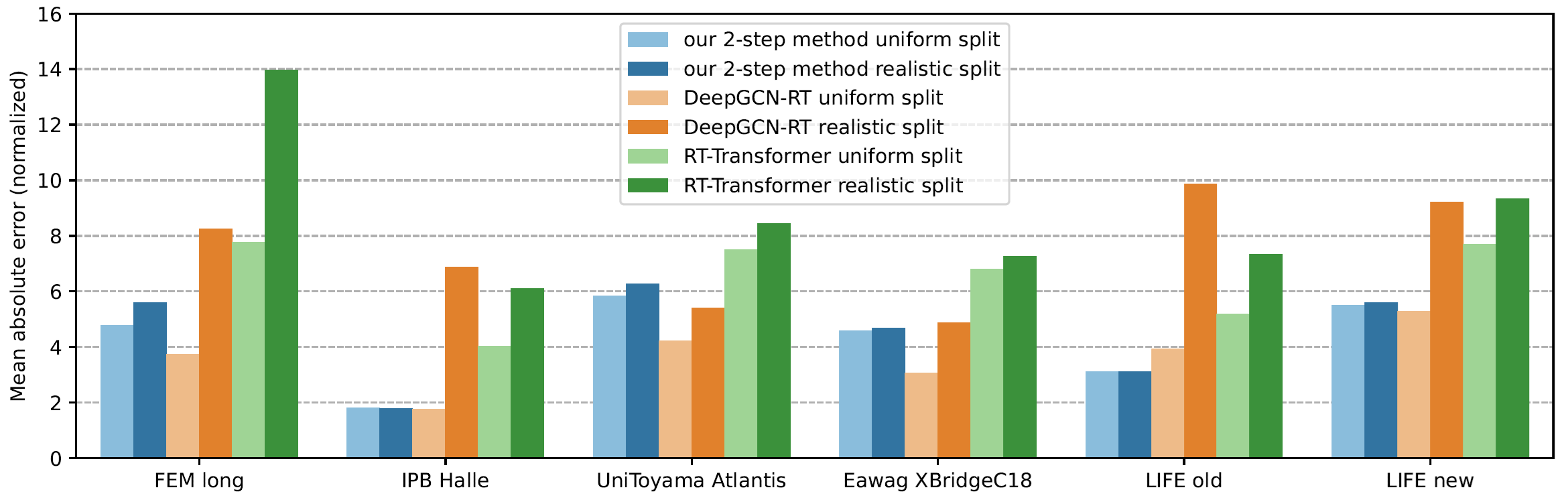
Why am I telling you that? I mean, besides showing off? (I sincerely hope that you share my somewhat twisted humor.) And, why did I say “already” above? Thing is, retention time prediction is dead, and you might not want to ruin your PhD student’s career by letting him/her try to develop yet another machine learning model for retention time prediction. But now, there is a new, much cooler task for you and/or your PhD student!
- If you are into graph machine learning, have a look at the problem of retention order index (ROI) prediction. This problem is both challenging and relevant, I can guarantee you that. Different from somewhat ill-posed problems of predicting complex biological traits such as toxicity, there is good reason to believe that ROI prediction can ultimately be “solved”, meaning that predictions become more accurate than experimental errors. On the other hand, ROI prediction is challenging, so complex & intricate models can demonstrate their power. Data are already available for thousands of compounds, trending upward. In conjunction with our 2-step approach, a better model for ROI prediction will automatically result in a better method for transferable retention time prediction. To give a ballpark estimate on the relevance: There are more than 2.1 million scientific studies that use liquid chromatography for the analysis of small molecules, and the chromatography market has an estimated yearly volume of 4 to 5 billion Euro. Your method may allow users to predict retention times for compounds that are illegal to have in the lab (be it tetrodoxin or cocaine), for all compounds in all spectral libraries and molecular structure databases combined, for compounds that do not even exist or which we presently do not know about.
- If you are doing LC experiments, you might want to upload your LC dataset with authentic standards to RepoRT. I would assume that our 2-step method (see below for details) will be integrated into many computational tools for compound annotation shortly; at least for one method I am sure. Alternatively, you might want to use 2-step to plan you next experiment, maximizing the separation of your compounds. Now, the 2-step method can truly do transferable prediction of retention times: It can do the predictions even for columns that are altogether missing from the training data. But things get easier and predictions get more accurate if there is a dataset in the training data with exactly your chromatographic setup. Let’s not make things overly complicated: A grandmaster in chess may be able to play ten games blindfolded and simultaneously, but s:he would not do so unless it is absolutely necessary. To this end, upload your reference datasets now, get better predictions in the very near future! It does not matter what compounds are in your dataset, as long as it contains a reasonable number (say, 100) of compounds. Everything helps; and in particular, it will help your own predictions for your chromatographic setup!
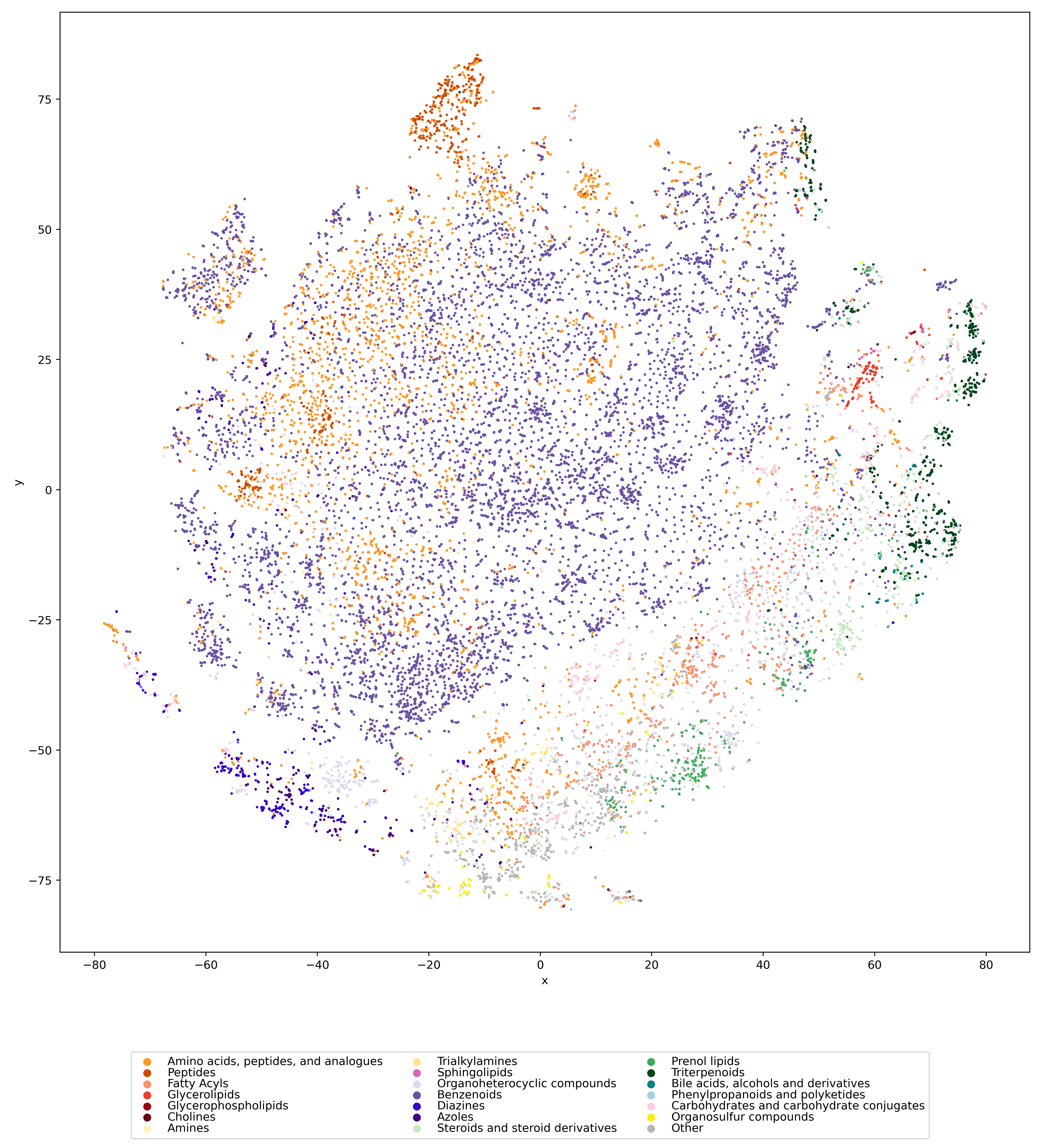
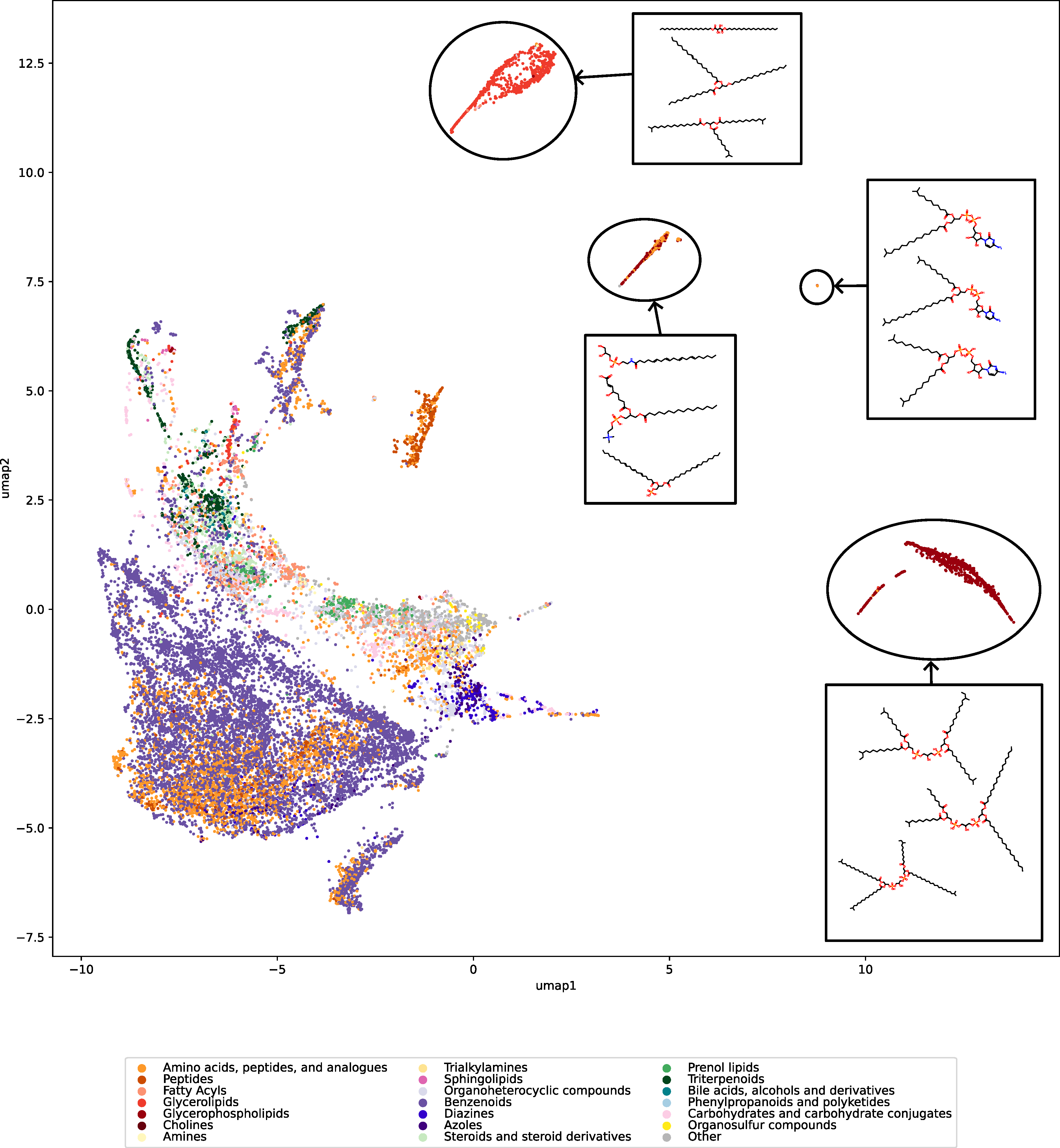
Now, how does the 2-step method work? The answer is: We do not try to predict retention times, because that is impossible. Instead, we train a machine learning model that predicts a retention order index. A retention order index (ROI) is simply a real-valued number, such as 0.123 or 99999. The model somehow integrates the chromatographic conditions, such as the used column and the mobile phase. Given two compounds A and B from some dataset (experiment) where A elutes before B, we ask the model to predict two number x (for compound A) and y (for compound B) such that x < y holds. If we have an arbitrary chromatographic setup, we use the predicted ROIs of compounds to decide in which order the compounds elute in the experiment. And that is already the end of the machine learning part. But I promised you transferable retention time, and now we are stuck with some lousy ROI? Thing is, we found that you can easily map ROIs to retention times. All you need is a low-degree polynomial, degree two was doing the trick for us. No fancy machine learning needed, good old curve fitting and median regression will do the trick. To establish the mapping, you need a few compounds where you know both the (predicted) ROI and the (measured) retention time. These may be authentic standards, such as the 19 NAPS molecules mentioned above. If you do not have standards, you can also use high-confidence spectral library hits. This can be an in-house library, but a public library such as MassBank will also work. Concentrate on the 30 best hits (cosine above 0.85, and 6+ matching peaks); even if half of these annotations are wrong, you may construct the mapping with almost no error. And voila, you can now predict retention times for all compounds, for exactly this chromatographic setup! Feed the compound into the ROI model, predict a ROI, map the ROI to a retention time, done. Read our preprint to learn all the details.
At this stage, you may want to stop reading, because the rest is gibberish; unimportant “historical” facts. But maybe, it is interesting; in fact, I found it somewhat funny. I mentioned above that I never wanted to work on retention time prediction. I would have loved if somebody else would have solved the problem, because I thought my group should stick with the computational analysis of mass spectrometry data from small molecules. (We are rather successful doing that.) But for a very long time, there was little progress. Yes, models got better using transfer learning (pretrain/fine-tune paradigm), DNNs and transformers; but that was neither particularly surprising nor particularly useful. Not surprising because it is an out-of-the-textbook application of transfer learning; not particularly useful because you still needed training data from the target system to make a single prediction. How should we ever integrate these models with CSI:FingerID?
In 2016, Eric Bach was doing his Master thesis and later, his PhD studies on the subject, under the supervision of Juho Rousu at Aalto University. At some stage, the three of us came up with the idea to consider retention order instead of retention times: Times were varying all over the place, but retention order seemed to be easier to handle. Initially, I thought that combinatorial optimization was the way to go, but I was wrong. Credit to whom credit is due: Juho had the great idea to predict retention oder instead of retention time. For that, Eric used a Ranking Support Vector Machine (RankSVM); this will be important later. This idea resulted in a paper at ECCB 2018 (machine learning and bioinformatics are all about conference publications) on how to predict retention order, and how doing so can (very slightly) improve small molecule annotation using MS/MS. (Eric and Juho wrote two more papers on the subject.)
The fact that Eric was using a RankSVM had an interesting consequence: The RankSVM is trained on pairs of compounds, but what it is actually predicting is a single real-valued number, for a given compound structure. For two compounds, we then compare the two predicted numbers; the smaller number tells you the compound that is eluting first. In fact, the machine learning field of learning-to-rank does basically the same, in order to decide which website best fits with your Google DuckDuckGo query. I must say that back then, I did not like this approach at all; it struck me as overly complicated. But after digesting it for some time, I had an idea: What if we do not use ROIs to decide which compounds elutes first? What if we instead map these ROIs to retention times, using non-linear regression? Unfortunately, I never got Eric interested to go into this direction. And I tried; yes I tried.
About seven years ago, Michael Witting and I rather reluctantly decided to jointly go after the problem. (I would not dare to go after a problem that requires massive chemical intuition without an expert; if you are a chemist, I hope you feel the same about applying machine learning.) We had my crazy idea of mapping ROIs to retention times, and a few backup plans in case this would fail. We had two unsuccessful attempts to secure funding from the Deutsche Forschungsgemeinschaft. My favorite sentence from the second round rejection is, “this project should have been supported in the previous round as the timing would have been better”; we all remember how the field of retention time prediction dramatically changed from 2018 to 2019…? But as they say, it is the timing, stupid! We finally secured funding in May 2019; and I could write, “and the rest is history”. But alas, no. Because the first thing we had to learn was that there was no (more precisely, not enough) training data. Jan Stanstrup had done a wonderful but painstaking job to manually collect datasets “from the literature” for his PredRet method. Yet, a lot of metadata were missing (these data are not required for PredRet, we must not complain) and we had to dig into the literature and data to close those gaps. Also, we clearly needed more data to train a machine learning model. With this, our hunt for more data began, which we partly scraped from publications, partly measured on our own; kudos go to Eva Harrieder! This took much, much longer than we would ever have expected, and resulted in the release of RepoRT in 2023. Well, after that, it is the usual. It works, it doesn’t work, maybe it works; next, it is all bad again; the other methods are so much better; the other methods have memory leakage; now, we have memory leakage; and finally, success. Phew… And here it is.
What about HILIC? I have no idea; but I can tell you that it is substantially more complicated. In fact, there currently is not even a description of columns comparable to HSM and Tanaka for RP. Maybe, somebody wants to get something started? Also, HILIC columns are much more diverse than RP columns, meaning that we need more training data (currently, we have less) and a better way to describe the column than for RP. But hey, start now so that it is ready in 10 years; worked for us!
References
- Kretschmer, F., Harrieder, E.-M., Witting, M., Böcker, S. Times are changing but order matters: Transferable prediction of small molecule liquid chromatography retention times. ChemRxiv, https://doi.org/10.26434/chemrxiv-2024-wd5j8-v3, 2024. Version 3 from August 2025.
- Bach, E., Szedmak, S., Brouard, C., Böcker, S. & Rousu, J. Liquid-Chromatography Retention Order Prediction for Metabolite Identification. Bioinformatics 34. Proc. of European Conference on Computational Biology (ECCB 2018), i875–i883 (2018). [DOI]
- Bach, E., Rogers, S., Williamson, J. & Rousu, J. Probabilistic framework for integration of mass spectrum and retention time information in small molecule identification. Bioinformatics 37, 1724–1731 (2021). [DOI]
- Bach, E., Schymanski, E. L. & Rousu, J. Joint structural annotation of small molecules using liquid chromatography retention order and tandem mass spectrometry data. Nat Mach Intel 4, 1224–1237 (2022). [DOI]
- Stanstrup, J., Neumann, S. & Vrhovšek, U. PredRet: prediction of retention time by direct mapping between multiple chromatographic systems. Anal Chem 87, 9421–9428 (2015). [DOI]
- Snyder, L. R., Dolan, J. W. & Carr, P. W. The hydrophobic-subtraction model of reversed-phase column selectivity. J Chromatogr A 1060, 77–116 (2004). [DOI]
- Kimata, K., Iwaguchi, K., Onishi, S., Jinno, K., Eksteen, R., Hosoya, K., Araki, M. & Tanaka, N. Chromatographic Characterization of Silica C18 Packing Materials. Correlation between a Preparation Method and Retention Behavior of Stationary Phase. J Chromatogr Sci 27, 721–728 (1989). [DOI]

 if we have to consider n objects. Again assuming that our quantum computer could do 1 billion such computations per second, we would end up with a running time of 1031 seconds, or 3.17 · 1023 years. That is still a looong time. There might be smarter ways of processing all structures on a quantum computer than using Grover’s algorithm; but so far, few such algorithms for even fewer problems have been found.
if we have to consider n objects. Again assuming that our quantum computer could do 1 billion such computations per second, we would end up with a running time of 1031 seconds, or 3.17 · 1023 years. That is still a looong time. There might be smarter ways of processing all structures on a quantum computer than using Grover’s algorithm; but so far, few such algorithms for even fewer problems have been found.
{kind=link}