At GCB 2024 (30th Sep. – 2nd Oct.), Fleming will give a presentation about RepoRT. Jonas, Leopold and Roberto will show their posters in the poster session.
News
Meet Sebastian at the SETAC Asia-Pacific Biennial Meeting in Tianjin
Sebastian will give a plenary talk at the SETAC Asia-Pacific 14th Biennial Meeting (September 21 to 25, 2024).
Meet Sebastian at the SMAP conference in Lille
Sebastian will give a plenary talk at the conference on Mass Spectrometry and Proteomic Analysis (SMAP), which will be held from September 16 to 19, 2024.
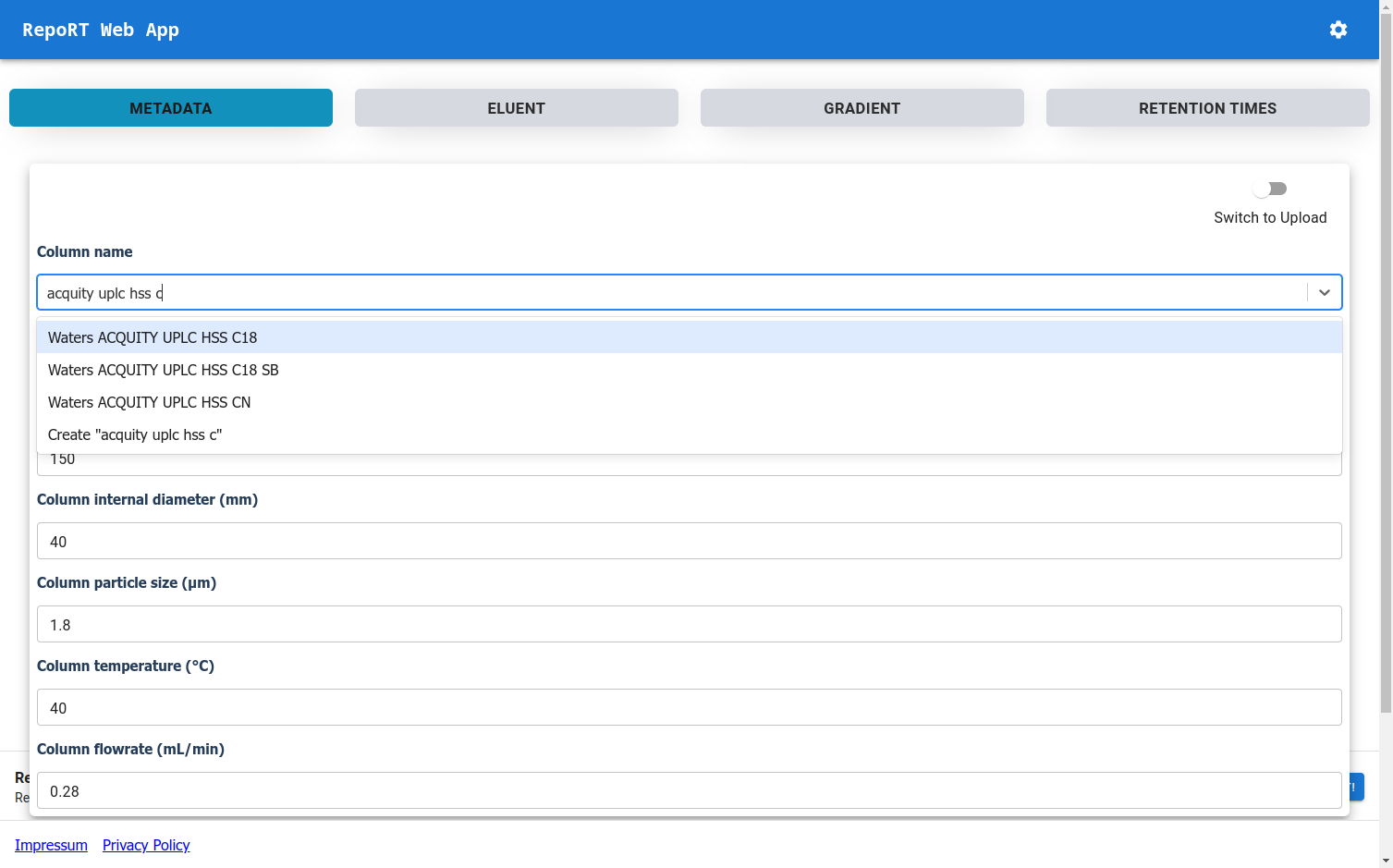
New Web-App for RepoRT
To make the submission process to RepoRT easier, we have launched a web app, available under https://rrt.boeckerlab.uni-jena.de. Submitters can upload their data without having to adhere to specific formatting requirements. Metadata and gradient information can be entered directly online. Part of the data validation happens during the uploading process, providing immediate feedback when something is wrong. Inside the web app, submitters can also keep track of all submissions and their current status. As we continue to work on improving the web app, your feedback is of course very welcome!
Meet Sebastian at the Okinawa Workshop for Computational Mass Spectrometry
On June 24, Sebastian will give a presentation on SIRIUS 6 at the Okinawa Workshop for Computational Mass Spectrometry.
Meet Jonas at the Computational Metabolomics Workshop in Aberdeen
At the Computational Mass Spectrometry Workshop (19-21 June 2024), Jonas will hold a presentation on SIRIUS 6.
Meet Sebastian, Kai and Fleming at the Metabolomics conference in Osaka
At the Metabolomics 2024 (16-20 June), Sebastian will give a keynote presentation. Kai and Fleming will hold a workshop on SIRIUS 6.
Meet Markus at the ASMS conference in Anaheim
Meet Markus at the ASMS conference 2024 in Anaheim between June 2-6!
Meet Andrés and Nils at the European School of Metabolomics in Granada
Meet us at the EUSM 2024 in Granada! Nils will give a hands-on session on SIRIUS.
Prague workshop will be streamed
Good news for those who want to attend the Prague workshop but were not admitted: The Prague workshop on computational mass spectrometry will be streamed, see here. You should get the necessary software installed beforehand if you want to join in.
IMPRS call for PhD student
The International Max Planck Research School at the Max Planck Institute for Chemical Ecology in Jena is looking for PhD students. One of the projects (Project 7) is from our group on “rethinking molecular networks”. Application deadline is April 19, 2024.
Mass spectrometry (MS) is the analytical platforms of choice for high-throughput screening of small molecules and untargeted metabolomics. Molecular networks were introduced in 2012 by the group of Pieter Dorrestein, and have found widespread application in untargeted metabolomics, natural products research and related areas. Molecular networking is basically a method of visualizing your data, based on the observation that similar tandem mass spectra (MS/MS) often correspond to compounds that are structurally similar. Constructing a molecular network allows us to propagate annotations through the network, and to annotate compounds for which no reference MS/MS data are available. Since its introduction, the computational method has received a few “updates”, including Feature-Based Molecular Networks and Ion Identity Molecular Networks. Yet, the fundamental idea of using the modified cosine to compare tandem mass spectra, has basically remained unchanged at the core of the method.
In this project, we want to “rethink molecular networks”, replacing the modified cosine by other measures of similarity, including fragmentation tree similarity, the Tanimoto similarity of the predicted fingerprints, and False Discovery Rate estimates. See the project description for details.
We are searching for a qualified and motivated candidate from bioinformatics, machine learning, cheminformatics and/or computer science who want to work in this exciting, quickly evolving interdisciplinary field. Please contact Sebastian Böcker in case of questions. Payment is 0.65 positions TV-L E13.
IMPRS: https://www.ice.mpg.de/129170/imprs
MPI-CE: https://www.ice.mpg.de/
SIRIUS & CSI:FingerID: https://bio.informatik.uni-jena.de/software/sirius/
Literature: https://bio.informatik.uni-jena.de/publications/ and https://bio.informatik.uni-jena.de/textbook-algoms/
Jena is a beautiful city and wine is grown in the region:
https://www.youtube.com/watch?v=DQPafhqkabc
https://www.google.com/search?q=jena&tbm=isch
https://www.study-in.de/en/discover-germany/german-cities/jena_26976.php
Prague workshop on computational MS overbooked
Unfortunately, the Prague Workshop on Computational Mass Spectrometry (April 15-17, 2024) is heavily overbooked. The organizers will try to stream the workshop and the recorded sessions will be made available online, so check there regularly.
The workshop is organized by Tomáš Pluskal and Robin Schmid (IOCB Prague). Marcus Ludwig (Bright Giant) and Sebastian will give a tutorial on SIRIUS, CANOPUS etc.
Topics of the workshop are: MZmine, SIRIUS, matchms, MS2Query, LOTUS, GNPS, MassQL, MASST, and Cytoscape.
Meet Sebastian at the DGMS conference in Freising
Meet Sebastian at the conference of the Deutsche Gesellschaft für Massenspektrometrie (DGMS 2024) in Freising! The conference is March 10-13, and Sebastian will give a keynote talk on Tuesday, March 12.
BTW, another keynote will be given by our close collaboration partner Michael Witting, also on March 12.
RepoRT has appeared in Nature Methods
Our paper “RepoRT: a comprehensive repository for small molecule retention times” has just appeared in Nature Methods. This is joint work with Michael Witting (Helmholtz Zentrum München) as part of the DFG project “Transferable retention time prediction for Liquid Chromatography-Mass Spectrometry-based metabolomics“. Congrats to Fleming, Michael and all co-authors! In case you do not have access to the paper, you can find the preprint here and a read-only version here.
RepoRT is a repository for retention times, that can be used for any computational method development towards retention time prediction. RepoRT contains data from diverse reference compounds measured on different columns with different parameters and in different labs. At present, RepoRT contains 373 datasets, 8809 unique compounds, and 88,325 retention time entries measured on 49 different chromatographic columns using varying eluents, flow rates, and temperatures. Access RepoRT here.
If you have measured a dataset with retention times of reference compounds (that is, you know all the compounds identities) then please, contribute! You can either upload it to GitHub yourself, or you can contact us in case you need help. In the near future, a web interface will become available that will make uploading data easier. There are a lot of data in RepoRT already, but don’t let that fool you; to reach a transferable prediction of retention time and order (see below), this can only be the start.
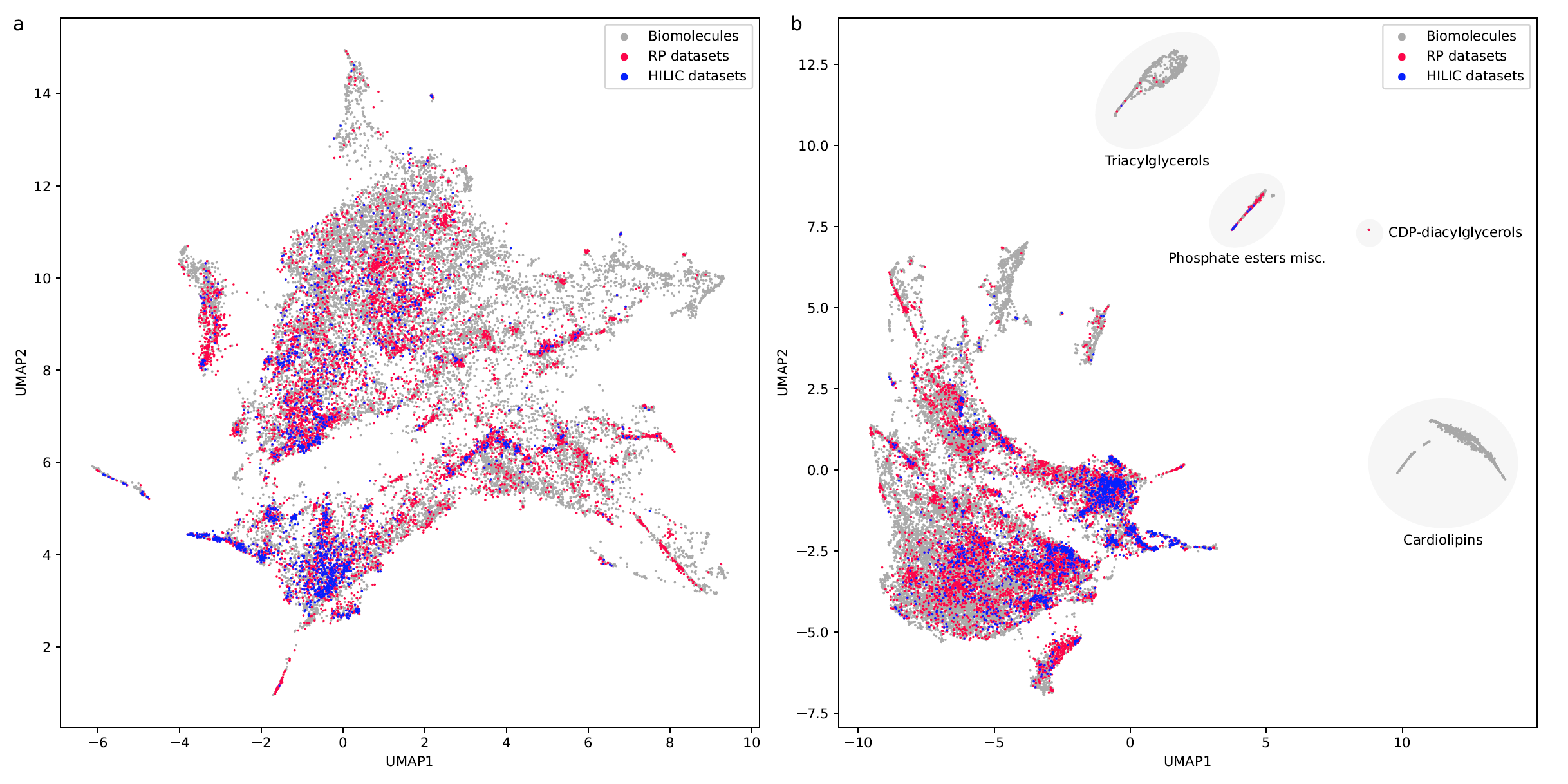
If you want to use RepoRT for machine learning and retention time or order prediction: We have done our best to curate RepoRT: We have searched and appended missing data and metadata; we have standardized data formats; we provide metadata in a form that is accessible to machine learning; etc. For example, we provide real-valued parameters (Tanaka, HSM) to describe the different column models, in a way that allows machine learning to transfer between different columns. Yet, be careful, as not all data are available for all compounds or datasets. For example, it is not possible to provide Tanaka parameters for all columns; please see the preprint on how you can work your way around this issue. Similarly, not all compounds that should have an isomeric SMILES, do have an isomeric SMILES; see again the preprint. If you observe any issues, please let us know. See this interesting blog post and this paper as well as our own preprint on why providing “clean data” as well as “good coverage” are so important issues for small molecule machine learning.
Bioinformatische Methoden in der Genomforschung muss leider ausfallen
Nach aktuellem Kenntnisstand muss das Modul “Bioinformatische Methoden in der Genomforschung” im WS 23/24 leider ausfallen. Wir dürfen die Mitarbeiterstelle nicht besetzen, die wir dafür zwingend brauchen. Wir haben gekämpft und argumentiert und alles getan was wir konnten, aber am Ende war es leider vergeblich. Das Modul findet voraussichtlich das nächste Mal im WS 25/26 statt.
Warnung: Im Zuge der Sparmaßnamen an der FSU Jena kann es in Zukunft häufiger zu sollen kurzfristigen Ausfällen kommen.
Meet Sebastian at the Munich Metabolomics Meeting
Sebastian will give a tutorial on using SIRIUS and beyond at the Munich Metabolomics Meeting 2023.
HUMAN EU PhD position is still open
Unfortunately, we have not been able to fill the PhD position for the HUMAN EU project so far. In case you are interested, please contact us!
Update: The position has been filled.
Neues Video zum Studium Bioinformatik
Im Rahmen des MINT Festivals in Jena hat Sebastian ein neues Video zum Studium der Bioinformatik aufgenommen: “Kleine Moleküle. Was uns tötet, was uns heilt“. Es richtet sich vom Vorwissen her an Schüler aus der Oberstufe, aber vielleicht können auch Schüler aus den Jahrgangsstufen darunter etwas mitnehmen. Das Video ist erst mal nur über diese Webseite zu erreichen, wird aber in Kürze bei YouTube hochgeladen.
Am Ende noch zwei Fragen: Erstens, welche Zelltypen im menschlichen Körper enthalten nicht die (ganze) DNA? Da war ich aus Zeitgründen bewusst schlampert; keine Regel in der Biologie ohne Ausnahme. Und zweitens, NMR Instumente werden häufig nicht mit flüssigem Stickstoff gekühlt, sondern mit… was? Wie so oft geht es dabei ums liebe Geld.
Meet Nils, Wei, Fleming and Sebastian at GCB 2023
Meet us at the GCB 2023 in Hamburg! Nils, Wei and Fleming are going to present posters, and Sebastian will give a keynote talk.
Meet Sebastian (remotely) and Fleming at the Swedish TB meeting
Sebastian and Fleming will participate in the Swedish National Tuberculosis meeting: Sebastian will give a talk remotely and Fleming will be on site in Umeå to give a hands-on session on SIRIUS.
